- Noticias / Una nueva variante en homocigosis en el péptido maduro IGF1 enfatiza la variabilidad de las manifestaciones clínicas y bioquímicas de los pacientes con deficiencia de IGF1
Comunicación de la ciencia: Noticia científica
Una nueva variante en homocigosis en el péptido maduro IGF1 enfatiza la variabilidad de las manifestaciones clínicas y bioquímicas de los pacientes con deficiencia de IGF1
Por Dra Ana Keselman y Dra Ayelen Martin
Compartir en
redes sociales
{kind=link}
Resumen
El retardo de crecimiento intrauterino es una entidad que involucra un grupo heterogéneo de pacientes con diversidad de diagnósticos. Las causas que pueden llevar a un paciente a no crecer adecuadamente intraútero pueden ser maternas, demográficas, placentarias y fetales. Dentro de las fetales, los defectos genéticos en los factores de crecimiento similares a la insulina (IGFs) pueden producir retardos de crecimiento pre y posnatales, muchas veces muy severos. Hasta el momento, sólo 3 pacientes portadores de variantes en el gen de IGF1 en homocigosis y caracterizadas funcionalmente han sido comunicadas en la bibliografía, con características clínicas y perfiles bioquímicos variables.
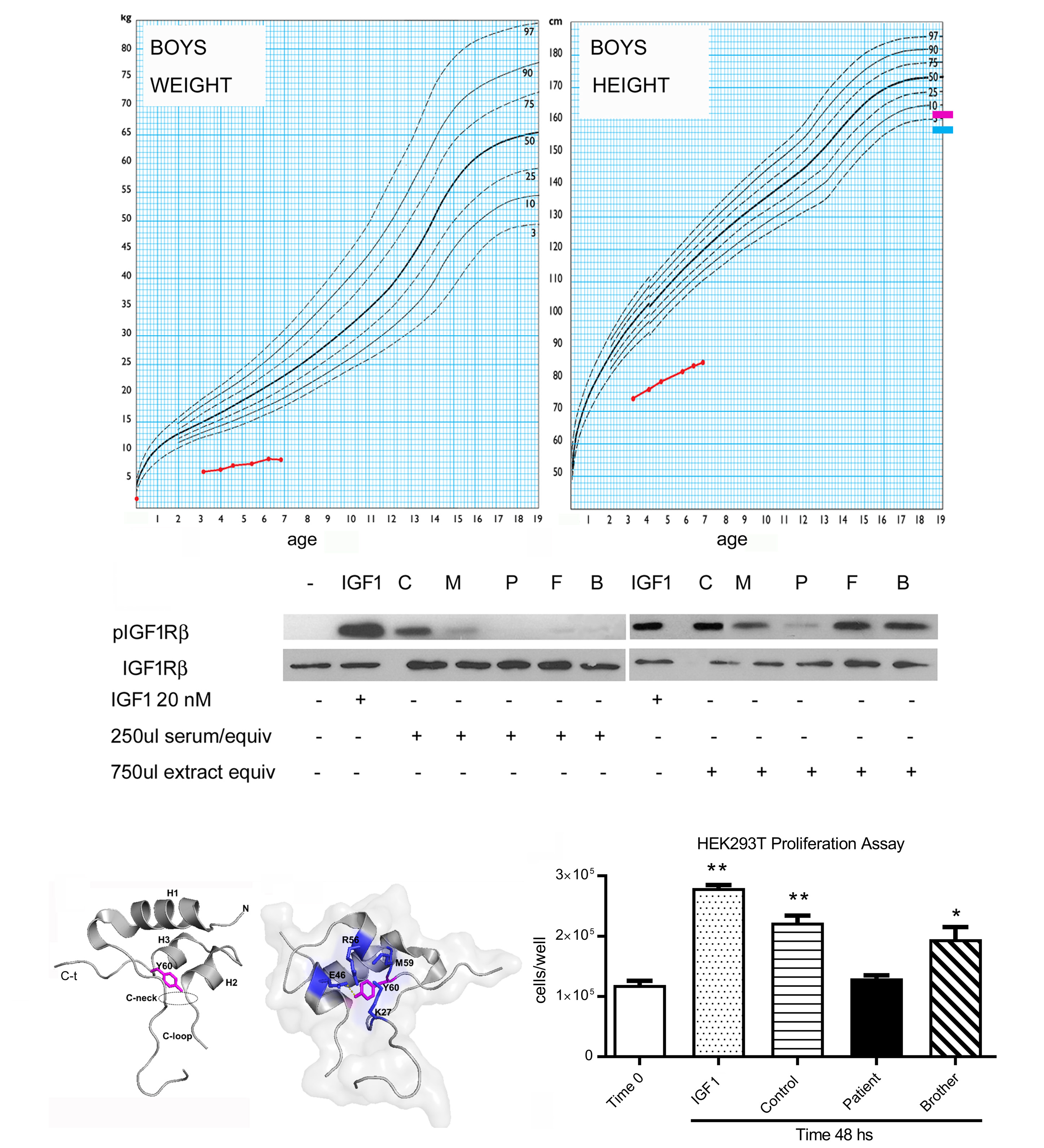
En este trabajo estudiamos un paciente con retardo de crecimiento intrauterino y baja talla severa, asociados a retraso madurativo, microcefalia, sordera neurosensorial, dismorfias y niveles variables de IGF1, con sospecha de insensibilidad a la hormona de crecimiento (GHI) secundaria a un defecto en el gen de IGF1. Luego de realizar estudios de genotipificación de nucleótido único (CGH-SNP array) asociado a secuenciación utilizando un panel clínico (NGS), se encontró una variante con cambio de sentido Tyr108His en el exón 3 del gen IGF1 en homocigosis. La variante Tyr108His, no descripta previamente, afecta a un aminoácido muy conservado localizado en el dominio A del IGF1, que es crítico para la interacción con su receptor (IGF1R). El estudio in sílico de esta variante utilizando diversas herramientas bioinformáticas la clasificó como patogénica de forma consistente (10/10 predictores de patogenicidad). El estudio de segregación alélica reveló que ambos progenitores y su medio hermano presentan la misma variante Tyr108His en heterocigosis. La caracterización funcional in vitro de esta variante consistió en la evaluación de la bioactividad de la variante de IGF1 utilizando muestras de suero del paciente, de controles sanos de la misma edad y de sus familiares. Para ello se realizaron estudios de activación (fosforilación) del IGF1R presente en la línea celular humana HEK293T luego de estímulos por un período corto, o luego de un período de 48 horas para evaluar el efecto sobre la proliferación celular. Pudimos comprobar que el suero del paciente no logró activar el IGF1R ni aumentar el número de células en cultivo, en contraste con los sueros de los controles, que activaron el IGF1R y duplicaron el número de células. Cabe destacar que, en ambos experimentos, los resultados obtenidos usando los sueros de los familiares heterocigotas fueron intermedios entre los del paciente y los controles.
El presente estudio contribuye al conocimiento, y enfatiza la variabilidad de las manifestaciones clínicas y bioquímicas debidas a mutaciones homocigotas en el gen de IGF1. El diagnóstico correcto de estos pacientes es fundamental para poder realizar las intervenciones adecuadas para la detección de comorbilidades, a la vez que contribuye a conocer los posibles tratamientos específicos.
Autoras: Keselman Ana & Martin Ayelen
Grupo Factores de Crecimiento y Biología Tumoral
Cita: A homozygous mutation in the highly conserved Tyr60 of the mature IGF1 peptide broadens the spectrum of IGF1 deficiency. Keselman AC*, Martin A*, Scaglia PA, Sanguineti NM, Armando R, Gutiérrez M, Braslavsky D, Ballerini MG, Ropelato MG, Ramirez L, Landi E, Domené S, Castro JF, Cassinelli H, Casali B, Del Rey G, Barros ÁC, Nevado Blanco J, Domené H, Jasper H, Arberas C, Rey RA, Lapunzina-Badía P, Bergadá I, Pennisi PA. Eur J Endocrinol. 2019 Nov;181(5):K43-K53. doi: 10.1530/EJE-19-0563.PMID: 31539878