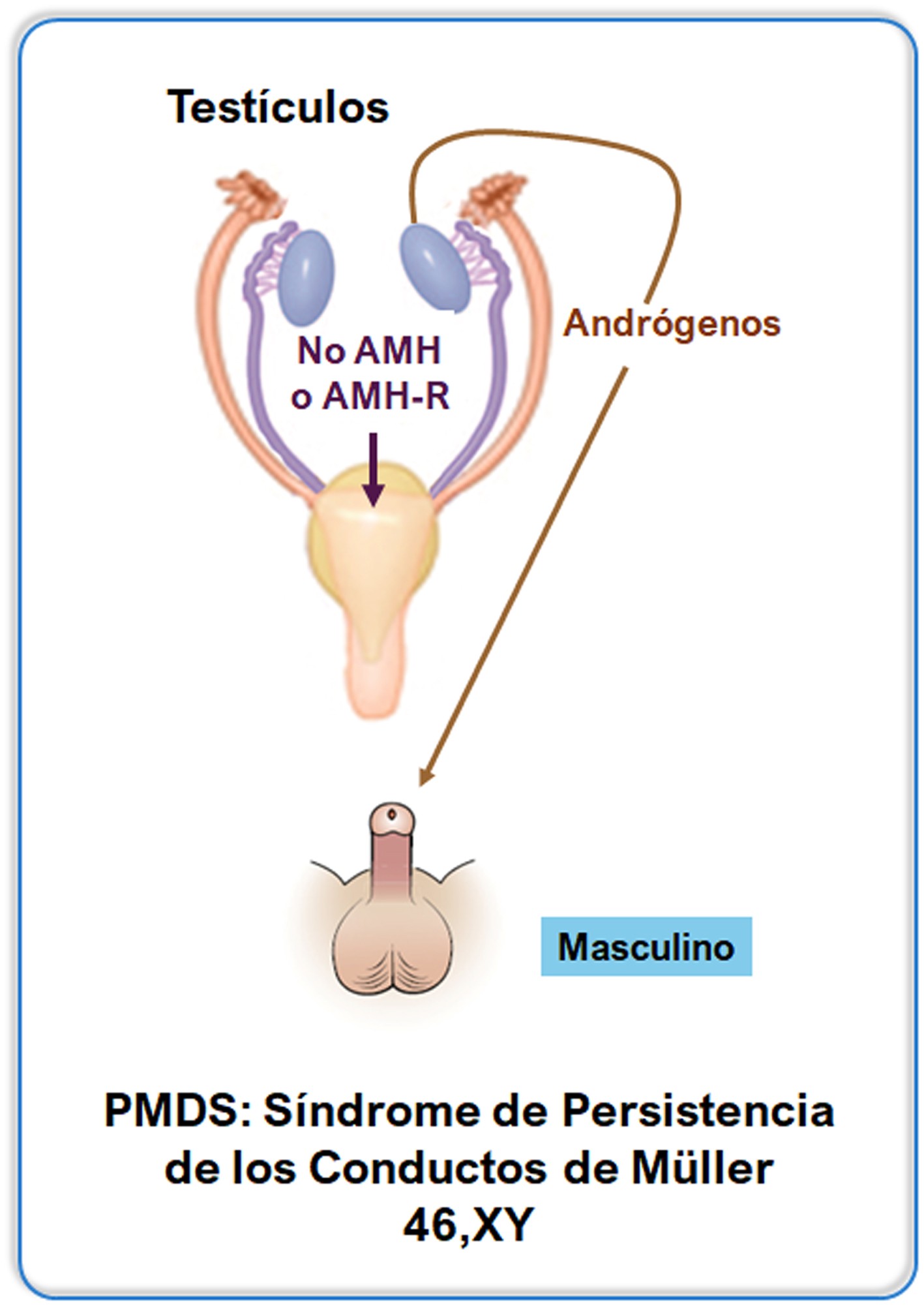
La hormona antimülleriana (AMH) provoca la regresión de los conductos de Müller en el feto masculino. La AMH se expresa en niveles altos en las células de Sertoli testiculares desde la vida fetal temprana hasta la pubertad. El gen humano de la AMH mapea en el cromosoma 19. El gen AMHR2, que codifica el receptor de AMH (AMH-R) tipo 2, se encuentra en el cromosoma 12. Las mutaciones en cualquiera de los genes son responsables del síndrome de persistencia de los conductos de Müller (PMDS), que se caracteriza por la presencia del útero y las trompas de Falopio en un varón que, por lo demás, está virilizado de forma normal. La AMH también es un biomarcador sérico útil de la función testicular en pacientes pediátricos.
Authors: Rey RA, Picard JY, di Clemente N, Cate RL, Josso N.
Cite: Anti-Müllerian hormone deficiency and resistance. In: Huhtaniemi I, Martini L, eds. Encyclopedia of Endocrine Diseases, 2nd Edition update. Oxford: Academic Press-Elsevier, 2024; Vol.5 pp 506-517, ISBN 978-0-12-812200-6. Reference Module in Biomedical Sciences, Elsevier, 2024, ISBN 9780128122006, https://doi.org/10.1016/B978-0-12-801238-3.65223-6.
Ver todas las Publicaciones Científicas del CEDIE
∼
Nuestras redes sociales:
Instagram: cedie.conicet
Sitio web Facebook: cedie.conicet
∼
{kind=link}